МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ РЕСПУБЛИКИ БЕЛАРУСЬ
БЕЛОРУССКИЙ ГОСУДАРСТВЕННЫЙ МЕДИЦИНСКИЙ УНИВЕРСИТЕТ
КАФЕДРА БИОЛОГИИ
Реферат
по медицинской биологии и генетике
на тему: «Гемоглобинопатии: серповидно-клеточная анемия и талассемия».
Минск 2015
Содержание
Оглавление
Введение. 3
Раздел 1. Серповидно-клеточная анемия. 4
Раздел 2. Талассемия. 10
Заключение. 15
Литература. 16
Введение
Гемоглобинопатия — наследственное или врождённое изменение или нарушение структуры белка гемоглобина, обычно приводящее к клинически или лабораторно наблюдаемым изменениям в его кислород-транспортирующей функции или в строении и функции эритроцитов.
К наиболее часто встречающимся и известным гемоглобинопатиям относятся серповидно-клеточная анемия, бета-талассемия.
Выделяют качественные гемоглобинопатии (изменения аминокислотной последовательности цепей глобина) и количественные гемоглобинопатии, или талассемии (снижение образования цепей глобина без изменения их структуры).
Раздел 1. Серповидно-клеточная анемия
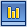
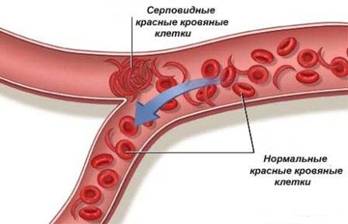
Серповидноклеточная анемия — это наследственная гемоглобинопатия, связанная с таким нарушением строения белка гемоглобина, при котором он приобретает особое кристаллическое строение — так называемый гемоглобин S. Эритроциты, несущие гемоглобин S вместо нормального гемоглобина А, под микроскопом имеют характерную серпообразную форму, за что эта форма гемоглобинопатии и получила название серповидно-клеточной анемии.
Серповидный эритроцит
Заболевание связано с мутацией гена HBB, вследствие чего синтезируется аномальный гемоглобин S, в молекуле которого вместо глутаминовой кислоты в шестом положении b-цепи находится валин. В условия гипоксии гемоглобин S полимеризуется и образует длинные тяжи, в результате чего эритроциты приобретают серповидную форму.
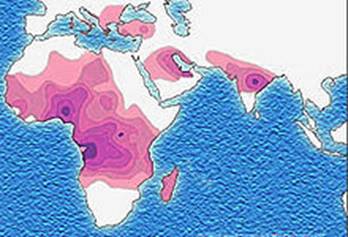
Серповидноклеточная анемия наследуется по аутосомно-рецессивному типу (с неполным доминированием). У носителей, гетерозиготных по гену серповидноклеточной анемии, в эритроцитах присутствуют примерно в равных количествах гемоглобин S и гемоглобин А. При этом в нормальных условиях у носителей симптомы практически никогда не возникают, и серповидные эритроциты выявляются случайно при лабораторном исследовании крови. Симптомы у носителей могут появиться при гипоксии (например, при подъёме в горы) или тяжёлой дегидратации организма. У гомозигот по гену серповидноклеточной анемии в крови имеются только эритроциты, несущие гемоглобин S, и болезнь протекает тяжело.
Эритроциты, несущие гемоглобин S, обладают пониженной стойкостью к лизису и пониженной кислород-транспортирующей способностью, поэтому у больных с серповидноклеточной анемией повышено разрушение эритроцитов в селезенке, укорочен срок их жизни, повышен гемолиз и часто имеются признаки хронической гипоксии (кислородной недостаточности) или хронического «перераздражения» эритроцитарного ростка костного мозга.
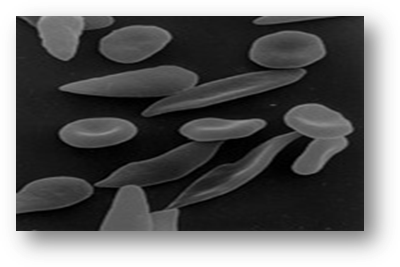
Серповидноклеточная анемия весьма распространена в регионах мира, эндемичных по малярии, причём больные серповидноклеточной анемией обладают повышенной (хотя и не абсолютной) врождённой устойчивостью к заражению различными штаммами малярийного плазмодия. Серповидные эритроциты этих больных также не поддаются заражению малярийным плазмодием в пробирке. Повышенной устойчивостью к малярии обладают и гетерозиготы-носители, которые анемией не болеют (преимущество гетерозигот), что объясняет высокую частоту этого вредного аллеля в африканских популяциях.
Распространение аллеля серповидно-клеточной анемии
Мутантный гемоглобин, вызывающий серповидно-клеточную анемию, препятствует использованию актинового цитоскелета малярийным плазмодием. При отсутствии заболевания из актиновых филаментов, протянутых под мембраной, плазмодий конструирует транспортную систему, с помощью которой отправляет наружу собственный белок адгезин. Этот адгезин, переброшенный на наружную сторону мембраны эритроцитов, делает клетки крови липкими. Эритроциты слипаются и оседают на стенках сосудов: это происходит, когда паразиту на очередном этапе жизненного цикла нужно выйти из кровотока. Процесс сопровождается множественными микрососудистыми воспалениями, характерными для малярии. Однако в клетках с серповидноклеточной мутацией плазмодию трудно заставить цитоскелет работать на себя, так как актиновый «мост» не дотягивается до мембранных везикул с адгезином, предназначенным для транспорта наружу. При постройке актинового «моста» плазмодий делает из коротких актиновых филаментов длинные, но дополнительная полимеризация актина невозможна в клетках с мутантным гемоглобином., поэтому носители гена серповидно-клеточной анемии устойчивы к малярии.
Симптомы
· Усталость и анемия
· Приступы боли
· Отек и воспаление пальцев рук и/или ног и артрит
·  Бактериальные инфекции
Бактериальные инфекции
· Тромбоз крови в селезенке и печени
· Лёгочные и сердечные травмы
· Язвы на ногах
· Асептический некроз
· Повреждение глаз
Образование тромба
Обычно новорождённые вполне здоровы, имеют нормальный вес и нормально развиваются, никаких симптомов у них не проявляется до 3-месячного возраста. Первыми признаками серповидноклеточной анемии у младенца обычно являются опухание и болезненность кистей рук или стоп, слабость и искривление конечностей и иногда, несколько позднее, отказ от ходьбы. Этот симптом является результатом закупорки эритроцитами капилляров мелких костей кистей и стоп и нарушения кровотока. Эритроциты выпадают из жидкой части крови и откладываются в капиллярах в виде осадка. Скопление эритроцитов постепенно рассасывается само по себе, но до тех пор, пока этого не произойдет, требуется помощь врача, чтобы смягчить боль и обнаружить возможные сопутствующие заболевания.
Единственным серьёзным осложнением серповидноклеточной анемии у ребёнка до 5-летнего возраста является инфекция. Скопление эритроцитов и закупорка капилляров в селезенке, органе, который в норме отфильтровывает бактерии из кровотока, происходит в течение первых лет жизни, что делает ребёнка особенно восприимчивым к смертельному заражению крови — сепсису. Поэтому родителей маленьких детей, страдающих серповидноклеточной анемией, предупреждают, чтобы они были внимательны и не пропустили ранних симптомов инфекции, таких как раздражительность, нервозность, повышенная температура и плохой аппетит. Если при заражении крови достаточно рано начинать применять антибиотики, фатальных осложнений можно избежать. После 5-летнего возраста, когда у ребёнка уже выработались соответствующие естественные антитела к такого рода бактериям, вероятность смертельной бактериальной инфекции существенно снижается.
Проблемой детей школьного возраста с серповидноклеточной анемией обычно является эпизодическая закупорка эритроцитами капилляров больших костей. Проявление: слабые ноющие боли в костях.
С возрастом процесс закупорки капилляров может затрагивать и другие органы. Например, в лёгких, развивается серьёзное респираторное заболевание. Очень редкое осложнение, которое бывает меньше чем у 10% больных с серповидноклеточной анемией — закупорка сосудов мозга, приводящая к инсульту.
У подростков отмечается задержка физического развития на 2-3 года. Однако со временем половая зрелость все же наступает, и исследования показывают, что женщины с серповидноклеточной анемией имеют нормальную возможность к деторождению.
У взрослых с серповидноклеточной анемией могут обнаруживаться симптомы хронической (постоянной или длительной) закупорки капилляров легких и почек, и может развиться хроническая легочная или почечная недостаточность.
Диагностика:
· общий анализ крови;
· биохимический анализ крови;
· электрофорез гемоглобина;
· ультразвуковое исследование (УЗИ);
· рентгенологическое исследование.
Профилактика:
Если один или оба родителя больны серповидноклеточной анемией, то их ребенок также может унаследовать данный недуг. Одним из методов, позволяющим определить вероятность наследования гена, ответственного за развитие данного заболевания, является полимеразная цепная реакция (ПЦР). Суть метода заключается в исследовании генетического материала обоих родителей и выявлении мутантных генов. При этом определяется как их наличие (или отсутствие), так и форма заболевания (гомозиготная или гетерозиготная).
Лечение: инфузионная терапия, обезболивание, антибактериальная терапия, заместительная терапия, препараты гидроксимочевины для активации синтеза HbF .
Раздел 2. Талассемия
Талассеми́я (анемия Кули) — заболевание, наследуемое по рецессивному типу (двухаллельная система), в основе которого лежит снижение синтеза полипептидных цепей, входящих в структуру нормального гемоглобина. В норме основным вариантом (97 %) гемоглобина взрослого человека является гемоглобин А. Это тетрамер, состоящий из двух мономеров α-цепей и двух мономеров β-цепей. 3% гемоглобина взрослых представлено гемоглобином А2, состоящем из двух альфа- и двух дельта-цепей. Существуют два гена HBA1 и HBA2, кодирующих мономер альфа, и один HBB-ген, кодирующий мономер бета. Наличие мутации в генах гемоглобина может привести к нарушению синтеза цепей определённого вида.
Альфа-талассемия
Связана с мутациями в генах HBA1 и HBA2. Есть всего 4 локуса, кодирующего α-цепи. Наличие мутации в одном из локусов приводит к минимальным клиническим проявлениям. Нарушения в двух локусах выражаются лёгкой формой анемии. При мутациях в трёх локусах возникает значительное уменьшение продукции α-глобина. При этом избыточные цепи β-глобина образуют тетрамеры— гемоглобин Н. Эта форма носит также название гемоглобинопатии Н. Характер заболевания может варьироваться от лёгкой до тяжёлой картины гипохромной микроцитарной анемии. Присутствие мутаций во всех четырёх аллелях альфа-глобина не совместимо с жизнью. Ребёнок с такой патологией погибает внутриутробно или вскоре после рождения. Из пуповинной крови таких детей можно выделить гемоглобин Барта.
Бета-талассемия
Существует два варианта бета-талассемии— большая талассемия CD8(-AA) и малая талассемия (minor), из которых большая талассемия — наиболее тяжёлая форма заболевания. Возникает при наличии мутаций в обоих аллелях гена бета-глобина. В отсутствие или при резком уменьшении производства бета-цепей гемоглобин А вытесняется гемоглобином F, в норме вырабатывающимся у плода и сменяющимся на гемоглобин А после родов. Малая талассемия связана с наличием мутации в одном из аллелей гена бета-глобина. Как правило, протекает легко и не требует лечения.
Этиология: талассемию вызывают точечные мутации или делеции в генах гемоглобина, ведущие к нарушению синтеза РНК, что приводит к уменьшению или полному прекращению синтеза одного из видов полипептидных цепей. Синтез цепей другого вида продолжается. Это приводит к образованию нестабильных полипептидных агрегатов из избыточных цепей, нарушающих нормальное функционирование эритроцитов и их разрушению. Повышенный гемолиз эритроцитов вызывает анемию.
Эпидемиология: альфа-талассемия распространена в Западной Африке и Южной Азии. Бета-талассемия часто встречается в странах Средиземноморья, Западной Азии и Северной Африки. Это регионы, где распространена малярия. Гетерозиготные носители мутаций в генах альфа- и бета цепей гемоглобина являются более устойчивыми к малярийному плазмодию. Имеются очаги талассемии в Азербайджане, в равнинных районах которого гетерозиготная бета-талассемия наблюдается у 7—10 % населения.
Диагностика:
· Общий осмотр: башенный череп, седловидная переносица, монголоидный разрез глаз, увеличение печени и селезенки, желтушность и бледность кожных покровов, язвы в области голеней, отставание в физическом и половом развитии.
· Анализ крови: снижение гемоглобина до 30-50 г/л, гипохромные эритроциты (красные клетки крови окрашены слабо вследствие низкого содержания в них гемоглобина), цветовой показатель (степень насыщения эритроцитов гемоглобином) 0,5 и ниже, увеличение ретикулоцитов (предшественники эритроцитов) до 2,5-4%, повышение железа сыворотки (жидкая часть) крови.
· Мазок крови: гипохромные эритроциты (слабо окрашенные) малых размеров (диаметр менее 7-8 мкм), мишеневидные (клетки с бледной тонкой периферией и центральным утолщением); характерен анизоцитоз (клетки разного размера) и пойкилоцитоз (изменение формы эритроцитов — от правильной, округлой, до овальной, серповидной и т.п.).
· Биохимия крови: гипербилирубинемия (повышение уровня билирубина за счет свободной фракции), гиперсидеремия (перегрузка железом), снижение общей железосвязывающей способности сыворотки (ОЖСС).
· Электрофорез гемоглобина на ацетат-целлюлозной пленке (pH 9,0 и 6,5) с последующим количественным определением гемоглобиновых фракций. При гомозиготной талассемии уровень фетального гемоглобина (гемоглобин плода; у взрослого человека в крови содержится лишь 1%) увеличен.
· Изучение биосинтеза цепей глобина in vitro (в пробирке).
· Пункция костного мозга: повышенное содержание сидеробластов (незрелые формы эритроцитов, которые имеют ядра).
· Рентгенологическое исследование костей: для большой β-талассемии характерны мелкие участки остеопороза (снижение плотности костной ткани) наряду с участками гипертрофии (увеличение объема и массы) костей черепа — так называемый симптом щетки или ежика, а также поперечная исчерченность мелких костей стоп и кистей.
· Молекулярное исследование (ПЦР), с помощью которого определяют мутацию в локусе β -глобина на 11-й паре хромосом, нарушающую синтез β -глобиновой цепи.
· Клинические и лабораторные данные при альфа-талассемии выражены менее отчетливо, чем при β-талассемии.
Лечение талассемии:
· При тяжелых формах (например, при большой β-талассемии) переливание цельной крови (временный эффект).
· В настоящее время наиболее эффективным считается переливание размороженных, отмытых или фильтрованных эритроцитов, которые гораздо реже вызывают побочные реакции, с одновременным длительным введением хелатов железа. При возникновении гемолитических кризов необходимо вводить глюкокортикоиды в небольших дозах.
· При больших размерах селезенки проводят ее удаление (спленоэктомия). Операцию не следует делать детям до 5 лет. Оптимальный возраст — 8-10 лет. Хороший эффект обычно наблюдается в течение первого года после удаления, затем снова возникает ухудшение. Также возрастает риск инфекционных заболеваний.
· В настоящее время наиболее предпочтительным считается пересадка (трансплантация) костного мозга. Это единственный метод радикального лечения талассемии. Однако найти подходящего донора обычно сложно.
· Больным следует соблюдать диету, употреблять продукты, которые содержат танин: чай, какао, а также орехи, сою. Эти продукты уменьшают всасывание железа.
Профилактика талассемии
· Первичная профилактика включает в себя пренатальную (дородовую) диагностику.
· Если отец и мать страдают талассемией, целесообразно исследование плода во время беременности на предмет заболевания талассемией с целью возможного своевременного прерывания беременности. Применяют два метода: фетоскопию и амниоцентез. С помощью них получают клетки плода (делают пункцию (прокол) через переднюю брюшную стенку, первый метод проводят под контролем УЗИ) и после проводят их медико-генетическое исследование. Данные методы считаются опасными, так как могут вызвать преждевременные роды, инфицирование и даже гибель плода.
· Родителям, у которых есть родственники с данной патологией, необходимо обратится до планирования беременности к генетику, чтобы тот назначил необходимое дородовое обследование.
Заключение
Генотерапия в будущем сможет значительно улучшить состояние или даже вылечить больных гемоглобинопатиями S, причем она не будет вызывать осложнений, характерных для аллотрансплантации. В начале 80-х гг. этот подход казался весьма перспективным. Но, если в лечении ферментопатий генотерапия добилась некоторых успехов, то при гемоглобинопатиях S трудностей гораздо больше. Так как болезнь вызвана экспрессией мутантного гена, простой перенос нормального гена в стволовую кроветворную клетку не препятствует образованию гемоглобина S . Кроме того, введенный ген должен работать чрезвычайно активно, так как в норме гемоглобина синтезируется на несколько порядков больше, чем других белков. Наконец, введенный ген должен экспрессироваться длительно, чего, как оказалось в экспериментах со стволовыми кроветворными клетками приматов, добиться нелегко.
Литература
1. https://ru.wikipedia.org/wiki/Гемоглобинопатии
2. https://ru.wikipedia.org/wiki/Серповидно-клеточная анемия
3. http://compulenta.computerra.ru/archive/biotechnology/645078/
4. http://www.polismed.com/articles-serpovidnokletochnaja-anemija-01.html
5. https://ru.wikipedia.org/wiki/Талассемия
6. http://lookmedbook.ru/disease/talassemiya
 (zip - application/zip)
(zip - application/zip)